Welcome
Welcome to the GenoPipe wiki!
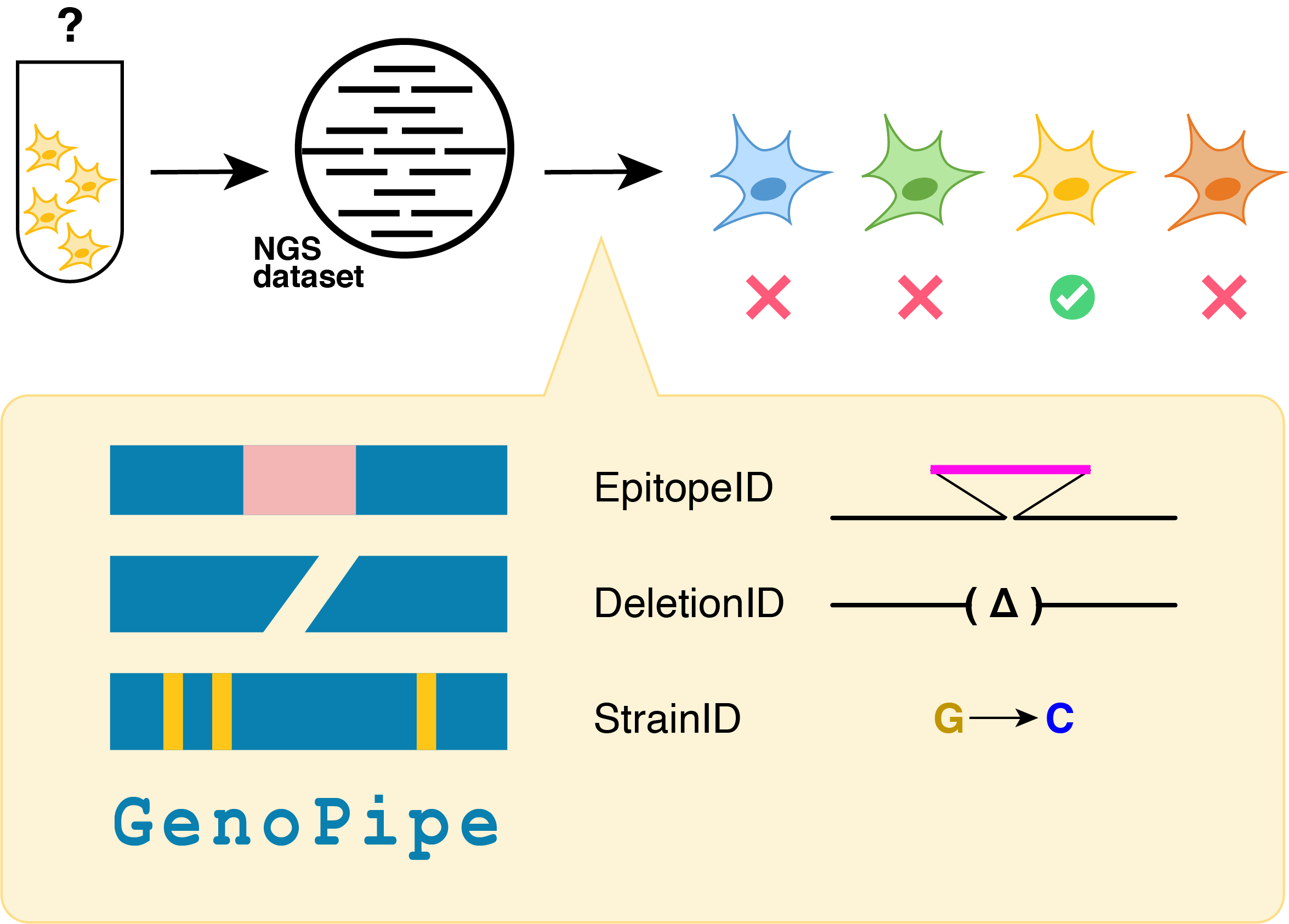
GenoPipe is a tool with three modules that check for different aspects of strain background in Next Generation Sequencing(NGS) datasets. Each module tests for a different aspect of strain background: epitope insertions, deletions, and cell line/variant-based background.
- EpitopeID identifies and determines the genomic location of epitopes or other inserted sequences relative to genomic loci.
- DeletionID identifies significant depletions of aligned NGS tags in the genome relative to a background model. This tool is set-up to confirm gene knockouts.
- StrainID compares a database of VCF files against an aligned BAM file to check for the presence of SNPs in order to determine a likely cell line/strain used in the experiment.
This is a great QC tool for your data fresh off the sequencer or for checking old published datasets when you can't find a record of the strain background used. These modules work for data from a variety of genomic sequencing assays, not just whole-genome sequencing (WGS) and can be customized to organisms other than yeast and human. Please check the docs and FAQs of the module you're interested in before starting. Confirm that your dataset and reference files are appropriate to use with the module.
System Requirements
Each GenoPipe module can run on Linux/MacOS systems. More available CPUs can allow the user to use the parallelization features for faster runtimes.
Genome Builds
Yeast (sacCer3) and human (hg19, hg38) genome builds supported across various modules "out-of-the-box"
Helper scripts for downloading reference genome sequence (FASTA) described in EpitopeID guide
- sacCer3 + scripts to rename chromosome identifiers to arabic numeral system
- hg19
Getting Help
- Check the specific module guides for a list of FAQs at the bottom. Pay attention to the tips and warnings
- For Bugs: please open an issue on Github with the following info
- command you ran with a description of the input files (we may ask you for a copy of the input files later)
- Entire error message or a description of the problem
- Software versions and OS you ran the command on
Citing Us
If you use GenoPipe in your work, you can use the BioRxiv publication (Lang et al, 2023).