Usage
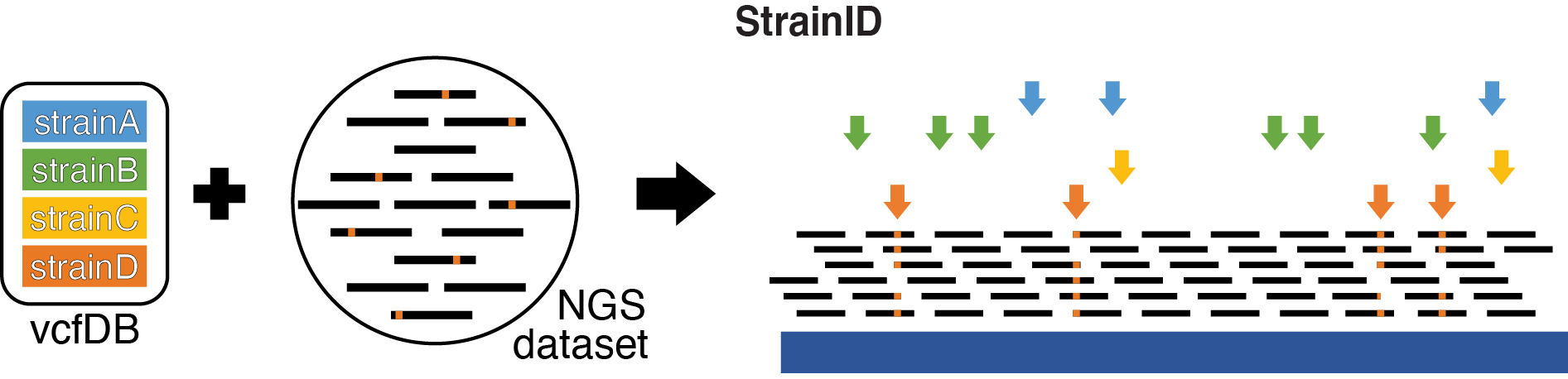
StrainID compares a database of VCF files against an aligned BAM file to check for the presence of SNPs in order to determine a likely cell line/strain used in the experiment.
bash identify-Strain.sh -i /path/to/BAM -g /path/to/genome/fASTA -v /path/to/VCF/files -o /path/to/output [ -s intSeed (default=None) ]
eg: bash identify-Strain.sh -i /input -g /sacCer3.fa -v /sacCer3_VCF -o /output -s 5
Dependencies
The following dependencies are needed to run StrainID.
- Anaconda
- Singularity
You can install these dependencies as a conda environment if you have conda setup on your machine using the following command:
conda create -n strainid-env -c bioconda -c conda-forge scipy pysam wget samtools
...and then turn on the environment with:
conda activate strainid-env
Definition files coming soon