Usage
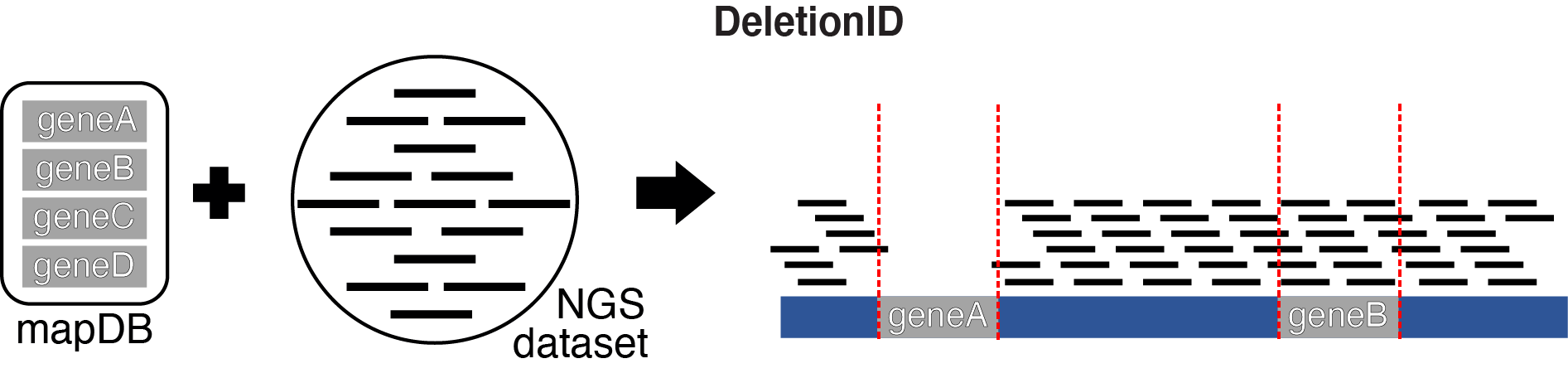
DeletionID identifies significant depletions of aligned NGS tags in the genome relative to a background model. This tool is set-up to confirm full gene knockouts.
bash identify-Deletion.sh -i /path/to/BAM -o /path/to/output -d /path/to/genome/database
eg: bash identify-Deletion.sh -i /input -o /output -d /sacCer3_Del
Dependencies
The following dependencies are needed to run DeletionID.
note
BedTools, BWA, and Perl are dependencies used by the utility scripts and are only needed for generating custom reference files. If you are working with the pregenerated set, you only need numpy, pysam, and samtools to
- Anaconda
- Singularity
You can install these dependencies as a conda environment if you have conda setup on your machine using the following command:
conda create -n deletionid-env -c bioconda -c conda-forge bedtools bwa perl numpy pysam samtools
...and then turn on the environment with:
conda activate deletionid-env
Definition files coming soon